
Deazaalloxazines – Flavin Derivatives That Provide Reductive Photoredox Catalysis with Inert Substrates
Abstract:
Reductive transformations of substances that are difficult to reduce continue to pose challenges for photoredox catalysis. Promising photoreduction catalysts include flavin and deazaflavin derivatives; however, even their reductive abilities are limited for the range of substrates considered “inert”. In this work, we present 5-deazaalloxazines, a new group of deazaflavin analogues that are predisposed to catalyze reductions due to their low reduction potential (down to −1.65 V vs. SCE) even in the ground state. We studied three series of 5-deazaalloxazines ([i] 5-unsubstituted, [ii] 5-aryldeazaalloxazines, and [iii] 5-trifluoromethyl-5-deazaalloxazines) to determine their photophysical and electrochemical properties and their ability to participate in model photoreduction reactions. From 31 compounds, we selected 1,3-dimethyl-7,8-dimethoxy-5-(o-tolyl)-5-deazaalloxazine [3a(o-MePh)], as it showed, among other things, the highest efficiency in photodehalogenation of p-fluoroanisole and was photostable and absorbed in the visible light region, thereby allowing photoreactions using a 400 nm LED. Practical applicability was demonstrated in the C─P coupling reaction of electron-rich aryl halides (including chloroanisoles and p-fluoroanisole) with trimethyl phosphite, providing an arylation reaction to form dimethyl arylphosphonates, and in the release/deprotection of amines from the corresponding tosyl and triflylamides.

7,8-Dimethoxy-5-aryl-5-deazaalloxazines (dAll) are new class of photostable, visible-light-absorbing flavin derivatives which are strongly reducible upon excitation with 400 nm light. It was demonstrated on difficult-to-reduce aryl halides like bromo-, chloro- and fluoroanisoles which were transformed to phosphonates by reductive cross-coupling with trimethyl phosphite or on desulfonylation of sulfonamides to free amines by S─N bond cleavage.
Redox properties of flavin derivatives: A comparative study
DOI:10.1016/j.electacta.2025.147520
Abstract:
Flavins as cofactors in natural systems as well as their artificial analogues used in catalysis are unique due to their specific redox properties. Our study provides the first direct comparison of the electrochemical behavior of various flavin derivatives: in addition to derivatives of the natural flavins possessing isoalloxazine structure, the experiments were oriented also to their analogues: alloxazines, deazaflavins and flavinium or deazaflavinium salts. We focused on the redox properties of six series of the mentioned flavin types with the stress on the substitution in positions 7 and 8 in connection with the structure of the core using various solvents and electrode materials. In addition to the study by cyclic and rotating disk voltammetry (CV and RDV), we turned our attention on the in-situ measurements of EPR spectra of radicals generated electrochemically. Experiments are accompanied by theoretical treatment enabling interpretation of electrochemical data and prediction of redox potential values of newly designed flavin derivatives.
Redox-innocent scandium(III) as the sole catalyst in visible light photooxidations
DOI:10.1038/s41467-025-63233-4
 Abstract:
Abstract:
In recent years, the catalytic activity of scandium triflate Sc(OTf)3 has attracted significant attention due to its robust Lewis acidity and the oxophilicity of Sc3+. These features have led to impressive progress in developing diverse organic reactions, including C-C bond formation. The Sc3+ also facilitates single electron transfer in photoinduced reactions either by coordination to an organophotoredox catalyst, which modifies its redox reactivity, or by the formation of a scandium–superoxide anion complex after electron transfer from a light-absorbing redox-active compound. The prior consideration of Sc3+ as a redox-inactive/innocent metal ion initially hampered the investigation of the possibility of using Sc(OTf)3 as a sole visible light photoredox catalyst. This work demonstrates the use of Sc(OTf)3 as a visible light photocatalyst capable of direct and mild aerobic oxidative C-H functionalisation of aromatic substrates by oxidation of the benzylic position and direct cyanation of the aromatic ring.
Deazaflavin-Catalyzed Arylation of White Phosphorus with Aryl Bromides and Chlorides
Abstract:
A substantial improvement in the challenging photocatalytic arylation of white phosphorus (P 4) with
aryl chlorides and bromides is reported. Using the readily accessible deazaflavin-based photocatalyst o-Me-dFl, valuable triarylphosphines (PAr 3) and tetraarylphosphonium salts ([PAr 4]X, X = Br, Cl) are synthesized from P4 under near UV-LED (365 nm) irradiation in up to 87% combined yield with drastically reduced reaction times compared to previous protocols. 31 P nuclear magnetic resonance spectroscopic monitoring studies and density functional theory calculations provide insights into the reaction pathway. The results represent an important step toward more atom-efficient and economical photocatalytic P4 functionalization reactions.
Introduction of flavin anions into photoredox catalysis: Acid-base equilibria of lumichrome allows photoreductions with an anion of an elusive 10-unsubstituted isoalloxazine
Abstract:
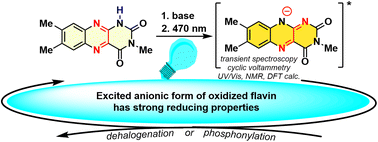 Flavins have been established as effective catalysts in oxidative photoredox catalysis. Conversely, their use in reductive photocatalysis remains limited, mainly due to the relatively low stability of the transient flavin radicals (semiquinones), which are used in photoreductions. The fully reduced forms of flavins are also disadvantaged in photocatalysis because they absorb light in the UV rather than in the visible region. In this work, we present a new approach for reductive flavin photocatalysis that utilises a flavin (isoalloxazine) anion derived from the elusive 10-unsubstituted 3,7,8-trimethylisoalloxazine, an unstable tautomer of 3-methyllumichrome. We found the conditions under which this isoalloxazine anion is formed by in situ deprotonation/isomerisation from the readily available 3-methyllumichrome and we subsequently used it as a photoredox catalyst in the reductive dehalogenation of activated bromoarenes and their C–P coupling reaction with trimethyl phosphite to form an arylphosphonate. Steady-state and transient absorption spectroscopy, NMR and cyclic voltammetry investigations, together with quantum chemical calculations, showed that the anion of oxidised isoalloxazine has several advantages, compared to other forms of flavins used in photoreductions, such as high stability, even in the presence of oxygen, an absorption maximum in the visible region, thereby allowing the use of excitation light between 470 and 505 nm, and a relatively long-lived singlet excited-state.
Flavins have been established as effective catalysts in oxidative photoredox catalysis. Conversely, their use in reductive photocatalysis remains limited, mainly due to the relatively low stability of the transient flavin radicals (semiquinones), which are used in photoreductions. The fully reduced forms of flavins are also disadvantaged in photocatalysis because they absorb light in the UV rather than in the visible region. In this work, we present a new approach for reductive flavin photocatalysis that utilises a flavin (isoalloxazine) anion derived from the elusive 10-unsubstituted 3,7,8-trimethylisoalloxazine, an unstable tautomer of 3-methyllumichrome. We found the conditions under which this isoalloxazine anion is formed by in situ deprotonation/isomerisation from the readily available 3-methyllumichrome and we subsequently used it as a photoredox catalyst in the reductive dehalogenation of activated bromoarenes and their C–P coupling reaction with trimethyl phosphite to form an arylphosphonate. Steady-state and transient absorption spectroscopy, NMR and cyclic voltammetry investigations, together with quantum chemical calculations, showed that the anion of oxidised isoalloxazine has several advantages, compared to other forms of flavins used in photoreductions, such as high stability, even in the presence of oxygen, an absorption maximum in the visible region, thereby allowing the use of excitation light between 470 and 505 nm, and a relatively long-lived singlet excited-state.
Cover picture: Chem. Sci., 2025, 16, 11681. DOI:10.1039/D5SC90144H
Divergent photoreduction of nitroaromatic compounds catalysed by dithienoquinoxaline
DOI:10.1016/j.jcat.2025.116033
Abstract:
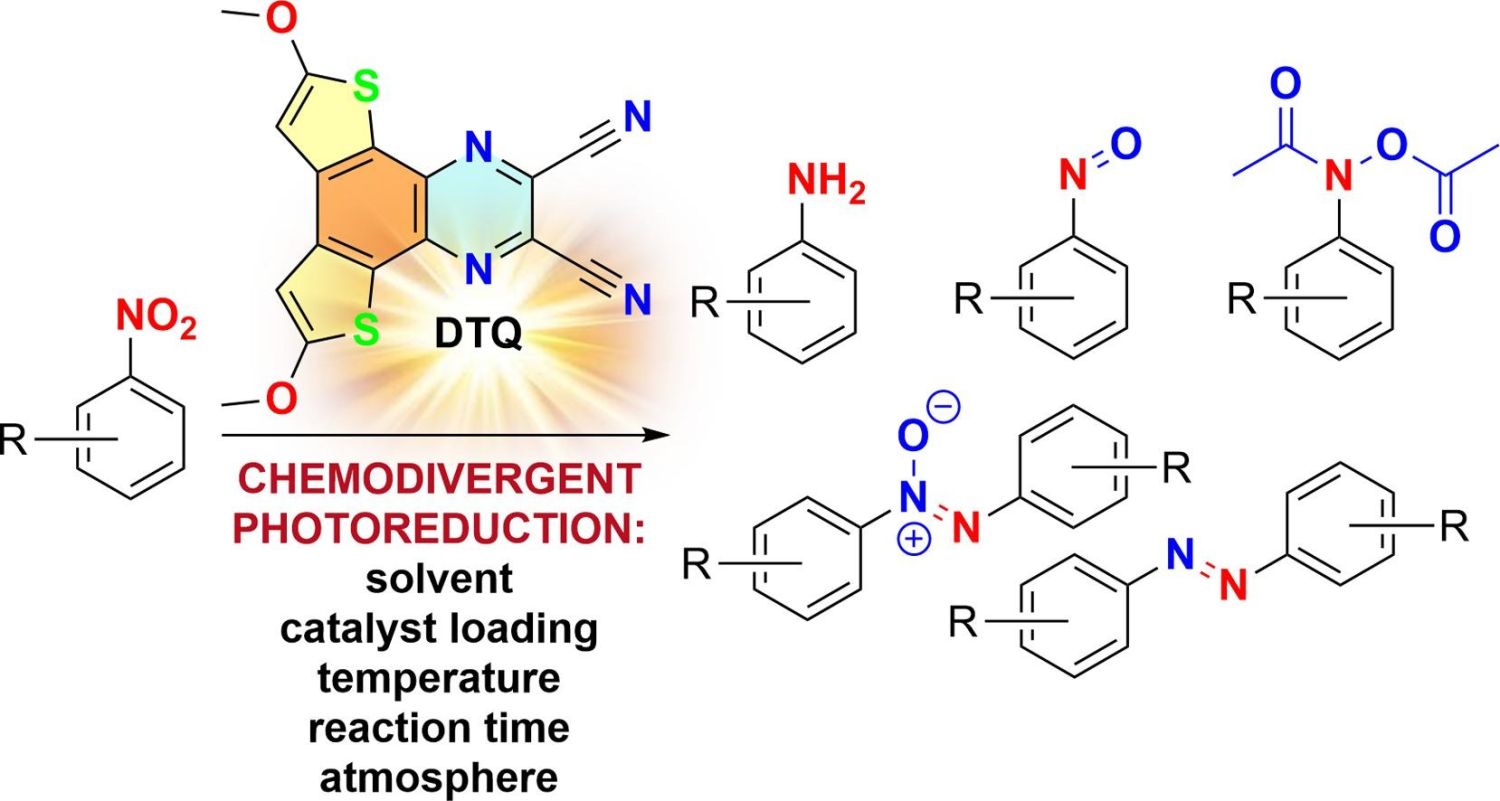 Nitroaromatic compounds can be conveniently reduced to anilines with the aid of a variety of chemical reductants. However, chemodivergent and chemoselective reduction of functionalized nitroaromatics into valuable nitroso, azo(xy), (hydr)azo and (hydroxyl)amino derivatives remains rather elusive. Electro- and photochemical strategies utilizing various catalytic systems and reaction media have been tested but most have been directed towards individual reduction products. Here, we present the photoredox-enabled chemodivergent reduction of nitroaromatics to nitroso, bis-(N,O-diacetyl)-N-arylhydroxylamine, azoxy, azo and amino derivatives. The developed protocol utilizes a novel photocatalyst, 6,9-dimethoxydithieno[2,3-f:3′,2′-h]quinoxaline-2,3-dicarbonitrile, and Hantzsch ester/triethanolamine as reductants. With the single organic photocatalyst, the reaction environment, temperature, time and catalyst loading can be varied to achieve chemodivergent photoreduction of nitroaromatics bearing various functional groups and to access a library of valuable products. The application of this methodology to multigram preparations of molecules of pharmaceutical and industrial relevance highlights its practical utility.
Nitroaromatic compounds can be conveniently reduced to anilines with the aid of a variety of chemical reductants. However, chemodivergent and chemoselective reduction of functionalized nitroaromatics into valuable nitroso, azo(xy), (hydr)azo and (hydroxyl)amino derivatives remains rather elusive. Electro- and photochemical strategies utilizing various catalytic systems and reaction media have been tested but most have been directed towards individual reduction products. Here, we present the photoredox-enabled chemodivergent reduction of nitroaromatics to nitroso, bis-(N,O-diacetyl)-N-arylhydroxylamine, azoxy, azo and amino derivatives. The developed protocol utilizes a novel photocatalyst, 6,9-dimethoxydithieno[2,3-f:3′,2′-h]quinoxaline-2,3-dicarbonitrile, and Hantzsch ester/triethanolamine as reductants. With the single organic photocatalyst, the reaction environment, temperature, time and catalyst loading can be varied to achieve chemodivergent photoreduction of nitroaromatics bearing various functional groups and to access a library of valuable products. The application of this methodology to multigram preparations of molecules of pharmaceutical and industrial relevance highlights its practical utility.
Biomimetic Orthogonal Removal of Arylacyl Protecting Groupsfrom the C-Terminus of Substituted Amino Acids andOligopeptides via Flavin N(5)-iminium Covalent Adducts
Abstract:
Flavin co-factors and other flavin derivatives are traditionally associated with one- or two-electron redox reactions. Flavins also form covalent intermediates with a variety of substrates. While extensive attention has focused on reactions at the C(4a) position of the flavin ring, recent interest has shifted toward the dynamic potential of the N(5) atom. In our study, we demonstrate the formation of artificial flavin N(5)-iminium covalent adducts via nucleophilic attack by enolates derived from arylacyl esters at the flavin N(5) position. This interaction mimics the biosynthesis of ether phospholipids catalysed by alkyl-dihydroxyacetone phosphate synthase via an N(5)-FAD iminium adduct formation and fatty acid release. Accordingly, we developed a method for the selective removal of phenacyl and 2-naphthylacetyl protecting groups from the C-terminus of α-amino acids and di- and tripeptides using biomimetic flavin organocatalysis. Although many approaches are currently available for protection of C-terminus of peptides, their removal often requires harsh conditions, which cause racemization and the loss of optical purity, together with other side reactions. Our method is versatile, mild, orthogonal to most functional and protecting groups and maintains optical purity of α-amino acids. From general point of view, the method signifies the groundbreaking application of the non-canonical reactivity of flavins in organic synthesis.
recognized by Synfacts (H. Yamamoto and T. Hattori: Synfacts 2025 Vol. 21 Issue 05 Pages 543-543, DOI: 10.1055/a-2558-1150)
Chemoselective Anaerobic Dehydrogenation of Alcohols by Using Visible Light and a Positively Charged Deazaflavin Catalyst Regenerated by Acetonitrile Solvent
Abstract:
Three series of novel deazaflavinum salts differing in their substitutions at positions 5 (R = H, phenyl, or mesityl), 7, and 8 (R = OMe, Me, H, or Cl) were synthesized as potential catalysts of a novel chemoselective visible light-mediated anaerobic oxidation of primary and secondary alcohols to carbonyl compounds. This mild procedure uses acetonitrile as a solvent, which acts simultaneously as a sacrificial electron acceptor (in place of the oxygen usually used in photooxidation reactions), and therefore the reaction does not need any additives. Structure and properties-versus-catalytic activity studies identified 5-mesityl-7,8-dimethoxy-3-methyldeazaflavinium chloride (3a-Cl) as the most potent catalyst. 3a-Cl was effective in non-deuterated acetonitrile (CH3CN), unlike its original 5-phenyl analogue 2a-Cl, which is efficient only in deuterated solvent (CD3CN). This difference arises because the regeneration of the 2a-Cl catalyst is slower in CH3CN than in CD3CN. Our method using the optimized 3a-Cl photocatalyst and CH3CN as a sacrificial oxidant and solvent in one is a useful addition to synthetic organic chemistry. Anaerobic conditions prevent side oxygenation reactions and overoxidations that usually occur in air or oxygen. This property makes this method suitable for dehydrogenations of alcohols that possess additional group(s) sensitive to oxygenation.
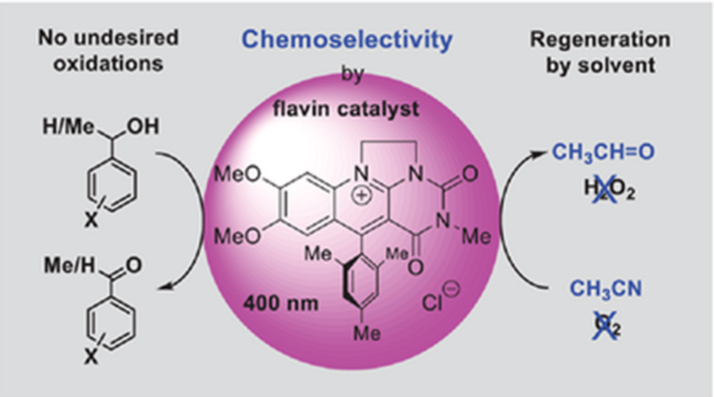
Chemoselective anaerobic dehydrogenation of primary and secondary alcohols to carbonyl compounds that leaves intact of additional oxygenation-sensitive functional groups is possible using a new flavin catalyst, 400 nm light, and acetonitrile, which acts simultaneously as a solvent and sacrificial oxidant. No other additives needed.
Fast singlet excited-state deactivation pathway of flavin with a trimethoxyphenyl derivative
DOI:10.1038/s41598-024-75239-x
Abstract:
Incorporation of the trimethoxyphenyl group at position 7 of flavin can drastically change the photophysical properties of flavin. We show unique fast singlet 1(π,π*) excited state deactivation pathway through nonadiabatic transition to the 1(n,π*) excited- state, and subsequent deactivation to the ground electronic state (S0), closing the photocycle. This mechanism explains the exceptionally weak fluorescence and the short excited–state lifetime for the flavin trimethoxyphenyl derivative and the lack of excited triplet T1 state formation. Full recovery of flavin in its ground state takes place within a 15 ps time window after photoexcitation in a polar solvent such as acetonitrile. According to quantum chemical calculations, the C(2)-O distance elongates by 0.16 Å in the 1(n,π*) state, with respect to the ground state. Intermediate–state structures are predicted by theoretical ab initio calculations and their dynamics are investigated using broadband vis-NIR time-resolved transient absorption and fluorescence up-conversion techniques.
A facile three-component route to powerful 5-aryldeazaalloxazine photocatalysts
Abstract:
Functionalized 5-aryldeazaalloxazines have been successfully synthesised through a one-pot, three-component reaction involving N,N-dimethylbarbituric acid, an aromatic aldehyde and aniline. By utilizing readily available reagents, this approach opens up the opportunity for the efficient formation of a variety of 5-aryldeazaalloxazines bearing electron-donating or halogen groups. This practical method is characterised by atom economy and offers a direct route to the introduction of an aryl moiety into the C(5)-position of deazaalloxazines, thereby generating novel catalysts for photoredox catalysis without the need for subsequent purification. Thus, it significantly improves existing approaches.

Ultrafast Events of Photoexcited Iron(III) Chloride for Activation of Benzylic C–H Bonds
DOI:10.1021/acs.jpclett.4c01116
Abstract: The usage of rare-earth-metal catalysts in the synthesis of organic compounds is widespread in chemical industries but is limited owing to its environmental and economic costs. However, recent studies indicate that abundant-earth metals like iron(III) chloride can photocatalyze diverse organic transformations using blue-light LEDs. Still, the underlying mechanism behind such activity is debatable and controversial, especially in the absence of ultrafast spectroscopic results. To address this urgent challenge, we performed femtosecond time-resolved electronic absorption spectroscopy experiments of iron(III) chloride in selected organic solvents relevant to its photocatalytic applications. Our results show that the long-lived species [Fe(II) ← Cl•]* is primarily responsible for both oxidizing the organic substrate and reducing molecular oxygen through the diffusion process, leading to the final product and regenerating the photocatalyst rather than the most widely proposed free chloride radical (Cl•). Our study will guide the rational design of efficient earth-abundant photocatalysts.
The usage of rare-earth-metal catalysts in the synthesis of organic compounds is widespread in chemical industries but is limited owing to its environmental and economic costs. However, recent studies indicate that abundant-earth metals like iron(III) chloride can photocatalyze diverse organic transformations using blue-light LEDs. Still, the underlying mechanism behind such activity is debatable and controversial, especially in the absence of ultrafast spectroscopic results. To address this urgent challenge, we performed femtosecond time-resolved electronic absorption spectroscopy experiments of iron(III) chloride in selected organic solvents relevant to its photocatalytic applications. Our results show that the long-lived species [Fe(II) ← Cl•]* is primarily responsible for both oxidizing the organic substrate and reducing molecular oxygen through the diffusion process, leading to the final product and regenerating the photocatalyst rather than the most widely proposed free chloride radical (Cl•). Our study will guide the rational design of efficient earth-abundant photocatalysts.
Catalyst-free aerobic photooxidation of sensitive benzylic alcohols with chemoselectivity controlled using DMSO as the solvent
Abstract:
Dicyanopyrazine photoredox catalysts: Correlation of efficiency with photophysics and electronic structure
DOI:10.1016/j.jcat.2024.115348
Abstract: Catalytic performance of three structurally-related dicyanopyrazine catalysts has been investigated in three photoredox transformations including deuteration of aldehydes, cross-coupling of iodo-substituted (hetero)aromatic substrates, and α-hydrogen abstraction from amines followed by annulation to pyrroloquinoline. Significantly different catalytic activity of the photocatalysts has been explained with the aid of electrochemical, spectroscopic, and quantum-chemical methods. Electrochemical measurements pointed to reversible one-electron reduction of the photocatalysts affording the corresponding radical anion, and, therefore, dicyanopyrazines are principally well-suited for reductive quenching cycle. Triplet excited state turned out to be a major excited species employed in photoinduced electron transfer. The measured excited state reduction potentials (Ered* = +1.88/+1.43 V) classify the (5–methoxy)thiophene-substituted dicyanopyrazines among the organic photocatalysts with high oxidation power, which is in contrast to N,N-dimethylanilino-substituted photocatalysts. Whereas 5–methoxythiophene photocatalyst forms triplet excited state almost independently on the solvent polarity, transient absorption spectroscopy evidenced the triplet state of N,N-dimethylanilino derivative only in nonpolar media. Moreover, its subsequent reduction to the corresponding radical anion is chemically cumbersome, which contrast to facile one-electron reduction of both cyano groups of photocatalyst bearing weak 5–methoxythiophene donors. The doublet excited radical anion of the latter proved to be very powerful but short-lived reductant with Eox* = –2.84 V. Its reduction power has been demonstrated in a cross-coupling reaction involving consecutive photoinduced electron transfer to preassociated iodo(hetero)arenes. Hence, bis(5-methoxythiophen-2-yl)-2,3-dicyanopyrazine can be utilized in photoredox catalysis either as powerful oxidant or reductant.
Catalytic performance of three structurally-related dicyanopyrazine catalysts has been investigated in three photoredox transformations including deuteration of aldehydes, cross-coupling of iodo-substituted (hetero)aromatic substrates, and α-hydrogen abstraction from amines followed by annulation to pyrroloquinoline. Significantly different catalytic activity of the photocatalysts has been explained with the aid of electrochemical, spectroscopic, and quantum-chemical methods. Electrochemical measurements pointed to reversible one-electron reduction of the photocatalysts affording the corresponding radical anion, and, therefore, dicyanopyrazines are principally well-suited for reductive quenching cycle. Triplet excited state turned out to be a major excited species employed in photoinduced electron transfer. The measured excited state reduction potentials (Ered* = +1.88/+1.43 V) classify the (5–methoxy)thiophene-substituted dicyanopyrazines among the organic photocatalysts with high oxidation power, which is in contrast to N,N-dimethylanilino-substituted photocatalysts. Whereas 5–methoxythiophene photocatalyst forms triplet excited state almost independently on the solvent polarity, transient absorption spectroscopy evidenced the triplet state of N,N-dimethylanilino derivative only in nonpolar media. Moreover, its subsequent reduction to the corresponding radical anion is chemically cumbersome, which contrast to facile one-electron reduction of both cyano groups of photocatalyst bearing weak 5–methoxythiophene donors. The doublet excited radical anion of the latter proved to be very powerful but short-lived reductant with Eox* = –2.84 V. Its reduction power has been demonstrated in a cross-coupling reaction involving consecutive photoinduced electron transfer to preassociated iodo(hetero)arenes. Hence, bis(5-methoxythiophen-2-yl)-2,3-dicyanopyrazine can be utilized in photoredox catalysis either as powerful oxidant or reductant.
Exploring the Reactivity of Flavins with Nucleophiles Using a Theoretical and Experimental Approach
Abstract:
Covalent adducts of flavin cofactors with nucleophiles play an important role in non-canonical function of flavoenzymes as well as in flavin-based catalysis. Herein, the interaction of flavin derivatives including substituted flavins (isoalloxazines), 1,10-ethylene-bridged flavinium salts, and non-substituted alloxazine and deazaflavin with selected nucleophiles was investigated using an experimental and computational approach. Triphenylphosphine or trimethylphosphine, 1-nitroethan-1-ide, and methoxide were selected as representatives of neutral soft, anionic soft, and hard nucleophiles, respectively. The interactions were investigated using UV/Vis and 1H NMR spectroscopy as well as by DFT calculations. The position of nucleophilic attack estimated using the calculated Gibbs free energy values was found to correspond with the experimental data, favouring the addition of phosphine and 1-nitroethan-1-ide into position N(5) and methoxide into position C(10a) of 1,10-ethylene-bridged flavinium salts. The calculated Gibbs free energy values were found to correlate with the experimental redox potentials of the flavin derivatives tested. These findings can be utilized as valuable tools for the design of artificial flavin-based catalytic systems or investigating the mechanism of flavoenzymes.

The ability of flavin derivatives to add a nucleophile can be quantified and the regioselectivity estimated by Gibbs free energy values calculated by DFT methods. The theoretical data corresponds to the results obtained by 1H NMR and UV/Vis spectroscopy and to experimental flavin reduction potentials.